|
|
Volume 69, pages 541-545, 1984
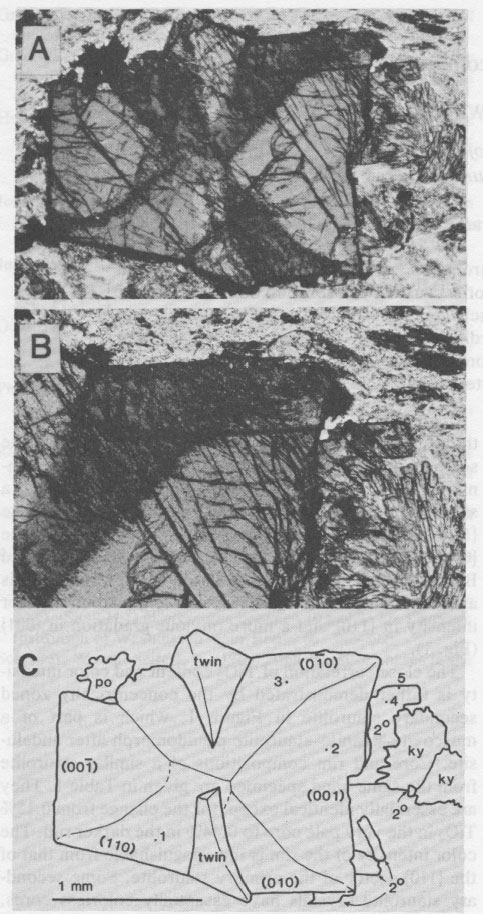
Titanium and the color of staurolite C. M. WARD Department of Geology University of Otago, Dunedin, New Zealand Abstract The visually assessed color intensity of staurolite appears to be directly proportional to the titanium content. It is inferred that the color is caused by absorption due to Fe2+-Ti4+ charge transfer. From this and the pleochroic scheme it is further inferred that the titanium in staurolite is located in the tetrahedrally coordinated Fe site. The marked difference in Ti content of the various sectors in sector-zoned staurolite can be rationalized more satisfactorily with Ti in this site than in the alternative octahedral Al site. Introduction Staurolite is well known for its yellow color and moderate pleochroism, which follows the scheme Z yellow, Y pale yellow, and X virtually colorless. It is commonly believed that Fe2+ in unusual tetrahedral coordination is responsible for the color (Ribbe, 1980), although Hollister and Bence (1967) suggested that Ti3+ and/or Fe3+ might be involved. Staurolite occurs in many different parageneses in a wide variety of pelites and metabasites in the Dusky Sound area of Fiordland, New Zealand. Data from these staurolites indicate a strong correlation between the titanium content of the staurolite and the color intensity. This correlation, the mechanism of color generation, and the inference of the structural site occupied by titanium, are the subjects of this paper. Correlation of color and titanium Figure 1 illustrates a strongly sector-zoned staurolite from a regional metamorphic terrane south of Dusky Sound. The different growth sectors {001}, {110} and {010} have distinctly different color intensities, with {010} >> {110} > {001}. The three sectors from a similar crystal in the same hand specimen were analyzed with the automated JEOL JXA-5A electron microprobe at the University of Otago, using a variety of natural and synthetic oxides and silicates as standards. The data were corrected on-line by the method of Bence and Albee (1968). Table 1 records the compositions of the different sectors based on 6-8 spot analyses in each sector at positions corresponding to 75-80% of the total linear growth. The analyses are very similar to the corresponding analyses by Hollister and Bence (1967) and Hollister (1970) of the sector-zoned staurolite from the Kwoiek area, British Columbia, in every relative sense, except that the differentiation of the sectors of the Dusky Sound staurolite is slightly less marked. Of particular interest is the excellent correlation between the TiO2 content of the sectors and their perceived color intensity. Careful scanning of staurolites from the Dusky Sound locality shows a slight increase in TiO2 content towards the rim of the {110} sector, and a major increase towards the rim of the {001} sector, as in the Kwoiek staurolites (Hollister and Bence, 1967, Fig. 3; and Hollister, 1970, Fig. 3). This again correlates with a just discernible gradation in color intensity in {110} and a more obvious gradation in {001} (Fig. 1). The close correlation of TiO2 content and color intensity is futher demonstrated by the concentrically zoned secondary staurolite in Figure 1, which is part of a muscovite-kyanite-staurolite pseudomorph after andalusite. Core and rim compositions of a similar staurolite from the same hand specimen are given in Table 1. They are essentially identical except for the change from 0.15% TiO2 in the very pale core to 0.44% in the darker rim. The color intensity of the rim is indistinguishable from that of the {l10} sector of the primary staurolite. Some secondary staurolite crystals have essentially colorless cores, and these contain TiO2 below the detection limit (~0.02%). The TiO2 contents of staurolites from a wide variety of rocks from the Dusky Sound area cover the complete range from <0.02% to 0.8%. Bearing in mind the importance of considering absorption only in the Z direction, and only in crystals occupying the full thickness of a standard thin section, there are no deviations detectable by eye from a relation of direct proportionality between color intensity and TiO2 content. There is only limited information in the literature with which to further assess the correlation of color intensity and titanium content, as the great majority of staurolite analyses record between 0.45 and 0.7% TiO2 and show little variation in color. In one noteworthy case, von Knorring et al. (1979) describe an unusually aluminous staurolite, with 9.3% FeO and no detectable TiO2, which is "very slightly pinkish with no pleochroism." Ganguly (1969) and Rao and Johannes (1979) synthesized Fe-staurolite from FeC2O4 • 2H2O, Al(OH)3 and SiO2 • nH2O, yet the resulting staurolite was observed to be yellow and pleochroic. This would appear to be a severe embarrassment to the hypothesis that titanium is required for the yellow color. However, the reagent grade reactants used are likely to have contained significant titanium. A sample of BDH reagent grade Al(OH)3 was found to contain 0.23% TiO2. Such an impurity level is probably normal, in view of the abundance of TiO2 in bauxite deposits and the general similarity of titanium and aluminum chemistry. Assuming quantitative reaction, staurolite with 0.19% TiO2 would be produced from this Al(OH)3. Such staurolite would be paler than usual but still distinctly yellow. Mechanism of color generation Identical polarized optical absorption spectra of staurolite have been obtained by Bancroft and Burns (1967) (see also Burns and Fyfe, 1967, and Burns, 1970) and Dickson and Smith (1976). There is a broad band centered about 1600 nm (6000 cm-1) in the infrared, and the tail (absorption edge) of a UV-centered peak in the blue end of the visible spectrum approaching 400 nm (25000 cm-1). This tail varies in size with vibration direction such that Z > Y > X, and is clearly responsible for the yellow color and pleochroism of staurolite. Burns and co-workers attributed the infrared absorption to spin-allowed d-d transitions in tetrahedrally coordinated Fe2+, and this has been supported by the success of molecular orbital calculations on FeIV by Vaughan et al. (1974) in matching the infrared spectrum. The UV peak is assumed to be due to oxygen-metal charge transfer, the metal being iron by implication. The extent of the visible tail of this peak must be largely unrelated to iron, however, because of the wide range of absorption intensities, down to essentially zero, in staurolites with rather constant FeO contents generally between 12 ½ and 14%. Hence consideration will be given to the role of titanium.
Having no 3d electrons, Ti4+ cannot by itself generate color by absorption due to d-d transitions, nor is O-Ti4+ charge transfer effective, since TiO2 is a well-known white pigment. Consequently it has in the past been commonly concluded that Ti3+ is responsible for the color of minerals where that color was correlated with titanium content, e.g., White and White (1967) for kyanite, Hollister and Bence (1967) for staurolite, and Burns and Fyfe (1967) and Manning and Nickel (1969) for titanaugite. However, Dowty and Clark (1973) concluded from study of an extraterrestrial "fassaite" extremely rich in Ti3+ (~10.7% Ti2O3) that absorption due to d-d transitions in Ti3+ was far too weak to account for the color of titanaugite, nor would it be appropriately polarized to produce the observed pleochroism. They suggested the alternative that heteronuclear Fe2+-Ti4+ charge transfer was responsible. This mechanism of color generation has since been widely reported, e.g., Parkin et al. (1977) in kyanite and Manning (1975) in vesuvianite. This latter study is of particular interest in that the vesuvianite concerned is closely analogous to staurolite, being yellow (correlating with titanium content) and pleochroic. The color is due to the super-imposition of polarized absorption bands in the vicinity of 23000 and 27000 cm-1 onto the tail of the UV-centered oxygen-metal charge transfer band. The hypothesis that Fe2+-Ti4+ charge transfer absorption is responsible for the yellow color and pleochroism of staurolite is the best available. Cation distribution Knowledge of the cation distribution in staurolite is necessary to further evaluate the possibility of Fe-Ti charge transfer, for only when Fe2+ and Ti4+ occupy adjacent sites will charge transfer be appreciable. Because of the structural complexities, particularly the partial occupancies of several sites, there is much uncertainty in cation site assignments. Considering first the titanium, with the exception of Smith (1968), all recent authors have assigned titanium to an octahedral site. There is, however, little evidence in support of this, and only Hollister (1970) has presented a case. He assigned Ti to the octahedral Al(3) sites on the basis of comparisons with the chemistry and structures of kyanite and spinels, in particular the relative preferences of the different cations for octahedral and tetrahedral sites in spinels. His analyses of the {001}, {110} and {010} sectors were interpreted in terms of substitution of Mg+Ti for 2Al in the Al(3) sites. The contrast in Ti content of the {001} and {101} sectors was explained by the ability of Mg and Ti to simultaneously enter adjacent Al(3) sites on the {010} faces (so maintaining local charge balance) and their inability to do so on {001}. Each of these three lines of support is suspect, however. First, Griffen and Ribbe (1973) showed that the structural analogy between staurolite and spinel is very crude and "conceptually invalid," and hence site preferences observed in spinel do not necessarily apply to staurolite. Second, the proposed substitution of Mg+Ti for 2Al, though not unreasonable, is far from quantitative in the sector-zoned staurolites and is likely to be a formalism only. Substitution patterns involving Ti may be much more complex, for example compare analyses 4 and 5, Table l, where the titanium content is to a large degree diffusion controlled in a chemical environment ("andalusite") initially heavily depleted in Fe, Mg and Ti. And third, Dowty (1976b) showed that simultaneous substitution of Mg+Ti onto a growing staurolite face was not necessary to maintain charge balance, because this would be more readily achieved by adjustment in the concentration of mobile anions adjacent to the growing face. In summary, there is no convincing positive evidence that titanium is present in an octahedral site.
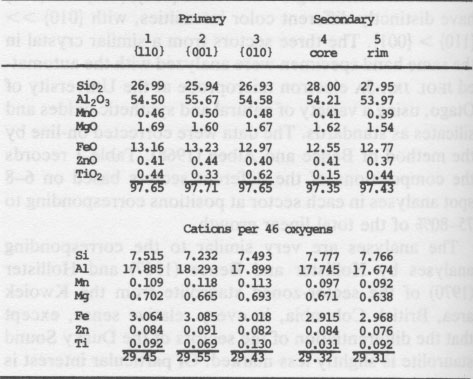
Table 1. Microprobe analyses of staurolite in OU 48699 Smith (1968) assigned cations to sites on the basis of a "horrendous mixture of experimental data and crystal-chemical speculation." His assignment of titanium to the Fe site is made without discussion. There is no convincing positive evidence that titanium is present in the tetrahedral site either. Considering now the iron, Smith (1968) assigned Fe2+ to the tetrahedral and octahedral sites (Al(3)+U) in the ratio of 3 to 1 on the basis of a Mössbauer spectrum. Dowty (1972) argued that the peaks attributed to octahedral iron were more probably due to another tetrahedral iron site, the two differing according to whether the adjacent U or Al(3) sites were occupied. However, after careful study of several natural staurolites and natural and synthetic spinels at low temperatures, Dickson and Smith (1976) reaffirmed the "octahedral" interpretation, though the evidence is not fully conclusive. Scorzelli et al. (1976) and Regnard (1976) add little to the discussion, and the latter's data are suspect in that the staurolite concerned has a rather unusual composition, with 40% Al2O3 and 45% SiO2. The site assignments made for iron by Smith (1968) are therefore accepted. The suggested iron and titanium sites all lie within the "iron" layer of the staurolite structure (Ward, 1984, Fig. 4), so that charge transfer is feasible with respect to interatomic distance, for all combinations of potential Fe and Ti sites. These combinations are as follows (with interatomic distances): model (l) Fe and Ti in adjacent Al(3A) and (3B) sites, 2.83Å; (2) Fe in the Fe site with Ti in Al(3A) or (3B), or vice versa, 3.40Å; (3) both Fe and Ti in Fe sites, 3.28Å. Combinations involving U sites are rejected because of their low occupancy, the excessive distance to the Al(3) sites (3.94Å), and the probability of adjacent Fe sites being empty when U sites are filled (Smith, 1968). Concerning combination (l), it might be argued that in view of the low total occupancy of these sites (~1/3) of which say 30% is Fe and 10% Ti, that the probability of Fe and Ti occupying adjacent individual sites is very low. However, a local charge balance mechanism may operate to greatly increase this probability, as postulated by Parkin et al. (1977) to account for the intensity of Fe2+-Ti4+ charge transfer absorption in kyanite with only trace FeO and TiO2 contents. Accepting the reality of the charge transfer, the character of the absorption can be used to discriminate between the possible cation site combinations. Manning (1975) clearly stated a very useful principle, that "intervalence charge transfer absorption is polarised accurately along metal-metal directions." This principle has been used with considerable confidence to deduce structures from spectral measurements (e.g., Manning, 1975, 1976; Dowty and Clark, 1973). Applied to staurolite with Z = c, Y = a and X = b, it implies that the Fe-Ti vector has a large component parallel to c, a smaller component parallel to a, and a near zero component parallel to b. Components of potential Fe-Ti vectors in a, b and c are as follows: model (1) 0, 0, 2.83Å; (2) 3.09, 0, 1.40; (3) 1.69, 0, 2.81. A zero component in b is consistent with all three site combinations, however the c > a not = 0 feature is specific to the third, i.e., both Fe and Ti in Fe sites. This is the first substantial evidence that titanium might be tetrahedrally coordinated in staurolite. Titanium sector-zoning re-examined The hypothesis that titanium occupies the tetrahedral Fe site rather than octahedral Al sites can be tested by its ability to account for the pattern of sector-zoning in titanium. Dowty (1976b), though discounting the bases of Hollister's (1970) model, could not satisfactorily explain the enrichment of Ti on {010} and its depletion on {001} relative to {110}. He (Dowty, 1976a,b) propounds a model of crystal growth based on the proposition that growth of each face takes place by the relatively rapid addition of unit slabs of thickness equivalent to the X-ray d spacing, with longer periods during which a critical surface, the surface crossed by the lowest total bond strength, is exposed to the matrix. During this time the cations (or vacant sites) at the surface (protosites of Nakamura, 1973) are susceptible to replacement by ions of smaller radius and/or higher charge, if the sites are suitably exposed. Figure 2 shows an [001] projection of the staurolite structure, with three alternative positions for the critical surface on (010). Surface (b), computed by Dowty and initially favored by him, requires preferential substitution of Ti into Al(2) positions. Surface (c), subsequently favored as that of least bonding by Dowty, requires preferential substitution of Ti into Al(1). Neither of these is satisfactory, since (001), which favorably exposes Al(1) and (3), (Dowty, 1976b, Fig. 5), preferentially takes up excess Al, when Ti should be preferred according to the model, especially in Al(3). A difficulty with Dowty's model is that there are two planes of sites or protosites on either side of the surface of least bonding, and which of them is on the "inside" is undefined. This point is very significant for planes of some symmetry types, illustrated by the (a) and (b) surfaces on (010) of staurolite. These surfaces are symmetrically equivalent and so are crossed by equal total bond strengths, yet they expose completely different protosites to the matrix (analogous to the two sides to a single surface where the direction of growth was not specified). Discrimination between these alternative positions can be made by using the principle that the critical surface, being that of greater longevity, will be the one of lower energy. This will clearly be surface (a) because of the lower charge density.
Staurolite [001] projection Fig. 2. [001] projection of staurolite structure. The "crystal" is imagined to be growing out from the bottom left corner. The heavier lines show "surfaces of least bonding" (Dowty 1976b) or surfaces of lowest energy on (110) and (010). These are exposed to the matrix for longer periods than other surfaces during growth of a staurolite crystal. Surfaces labeled a, b and c on (010) are alternative positions for the critical surface (see text).
The outstanding feature of surface (a) is the high concentration of exposed Fe protosites, 4.5 per square nanometer, compared with 1.9 on the (110) surface of least bonding (Fig. 2). Fe sites are not exposed on the (001) surface (Dowty, 1976b, Fig. 5). Thus the predicted concentration of Fe protosites exposed on the critical surfaces, or more specifically the length of time these protosites are exposed to the matrix on the different faces, correlates very well with the observed TiO2 concentrations. Sector-zoning with respect to titanium can be rationalized more successfully if Ti4+ is taken to be in the tetrahedral Fe site rather than the octahedral Al(3) sites. Conclusions Iron cannot be primarily responsible for the yellow color of staurolite, as there is a wide variation from colorless to moderately intense yellow color in staurolites with fairly constant FeO contents usually in the range 12%-14%. Instead, the visually assessed color intensity appears to be directly proportional to the titanium content: normal yellow staurolites have about 0.5% TiO2, some more intensely yellow ones have up to 0.8% TiO2, and TiO2 is undetected in rare colorless staurolite. The most likely color-generating mechanism is Fe2+-Ti4+ charge transfer. The orientation of the Fe-Ti vector, deduced from the pleochroic scheme, indicates that the titanium is located in the tetrahedrally coordinated Fe site. However, it would be uncharacteristic for staurolite if Ti were restricted exclusively to this site. If significant Ti occupies other sites and if the relative extent of these substitutions is variable, then slightly different pleochroic schemes and/or anomalously pale staurolites might result. This is probably uncommon; staurolites from Dusky Sound which at first glance have appeared to be anomalously pale for their TiO2 content have proven to be either sections far from parallel to Z, or very fine staurolites considerably thinner than 30 µm. The pattern of TiO2 contents in the different sectors of sector-zoned staurolite can be rationalized readily when Ti is taken to be in the Fe site, whereas previous models in which Ti was assumed to occupy an octahedral Al site have been unsatisfactory. With this independent support, the case for the assignment of Ti to the tetrahedrally coordinated Fe site is strong. Acknowledgments Drs. A. F. Cooper, A. Reay and D. S. Coombs made useful criticisms of the manuscript. Dr. Reay kindly analysed the Al(OH)3 for titanium. Dr. Y. Kawachi gave instruction and advice on the use of the microprobe. I am grateful for all this assistance. References Bancroft, G. M. and Burns, R. G. (1967) Interpretation of the electronic spectra of iron in pyroxenes. American Mineralogist, 52, 1278-1287. Bence, A. E. and Albee, A. L. (1968) Empirical correction factors for the electron microanalysis of silicates and oxides. Journal of Geology, 76, 382-403. Burns, R. G. (1970) Mineralogical Applications of Crystal Field Theory. Cambridge University Press, Cambridge. Burns, R. G. and Fyfe, W. S. (1967) Crystal field theory and the geochemistry of transition elements. In R. H. Abelson, Ed., Researches in Geochemistry Vol. II, p. 259-285. Wiley, New York. Dickson, B. L. and Smith, G. (1976) Low temperature optical absorption and Mössbauer spectra of staurolite and spinel. Canadian Mineralogist, 14, 206-215. Dowty, E. (1972) Site distribution of iron in staurolite. Earth and Planetary Science Letters, 15, 72-74. Dowty, E. (1976a) Crystal structure and crystal growth: I. The influence of internal structure on morphology. American Mineralogist, 61, 448-459. Dowty, E. (1976b) Crystal structure and crystal growth: II. Sector zoning in minerals. American Mineralogist, 61, 460469. Dowty, E. and Clark, J. R. (1973) Crystal structure refinement and optical properties of a Ti3+ fassaite from the Allende meteorite. American Mineralogist, 58, 230-242. Ganguly, J. G. (1969) Chloritoid stability and related parageneses: theory, experiment and applications. Journal of Petrology, 13, 335-365. Griffen, D. T. and Ribbe, P. H. (1973) The crystal chemistry of staurolite. American Journal of Science, 273A, 479-495. Hollister, L. S. (1970) Origin, mechanism and consequences of compositional sector-zoning in staurolite. American Mineralogist, 55, 742-766. Hollister, L. S. and Bence, A. E. (1967) Staurolite: sectoral compositional variations. Science, 158, 1053-1056. Manning, P. G. (1975) Charge-transfer processes and the origin of colour and pleochroism of some titanium-rich vesuvianites. Canadian Mineralogist, 13, 110-116. Manning, P. G. (1976) Ferrous-ferric interaction on adjacent face-sharing anti-prismatic sites in vesuvianites: evidence for ferric iron in eight coordination. Canadian Mineralogist, 14, 216-220. Manning P. G. and Nickel, E. H. (1969) A spectral study of the origin of colour and pleochroism of a titanaugite from Kaiserstuhl and of a riebeckite from St. Peter's Dome, Colorado. Canadian Mineralogist, 10, 71-83. Nakamura, Y. (1973) Origin of sector-zoning in igneous pyroxenes. American Mineralogist, 58, 986-990. Parkin, K. M., Loeffler, B. M. and Burns, R. G. (1977) Mössbauer spectra of kyanite, aquamarine, and cordierite showing intervalence charge transfer. Physics and Chemistry of Minerals, 1, 301-311. Rao, B. B. and Johannes, W. (1979) Further data on the stability of staurolite + quartz and related assemblages. Neues Jahrbuch für Mineralogie, Monatshefte, 437-447. Regnard, J. R. (1976) Mössbauer study of natural crystals of staurolite. Journale de Physique Colloque, 37, C6, 797-800. Ribbe, P. H. (1980) Staurolite. In P. H. Ribbe, Ed., MSA Reviews in Mineralogy, v. 6: Orthosilicates. Mineralogical Society of America, Washington, D.C. Scorzelli, R. B., Baggio-Saitovich, E., and Danon, J., (1976) Mössbauer spectra and electron exchange in tourmaline and staurolite. Journale de Physique Colloque 37, C6, 801-805. Smith, J. V. (1968) The crystal structure of staurolite. American Mineralogist, 53, 1139-1155. Vaughan, D. J., Tossell, J. A. and Johnson, K. H. (1974) The bonding of ferrous iron to sulfur and oxygen in tetrahedral coordination: A comparative study using SCF Xα scattered wave molecular orbital calculations. Geochimica et Cosmochimica Acta, 38, 993-1005. von Knorring, O., Sahama, T. G. and Siivola, J. (1979) Zincian staurolite from Uganda. Mineralogical Magazine, 43, 446. Ward, C. M. (1984) Magnesium staurolite and green chromian staurolite from Fiordland, New Zealand. American Mineralogist, 69, 531-540. White, E. W. and White, W. B. (1967) Electron microprobe and optical absorption study of colored kyanites. Science, 158, 915-917. Manuscript received, December 28, 1982; accepted for publication, September 27, 1983. [var:'startyear'='1984'] [Include:'footer.htm'] |